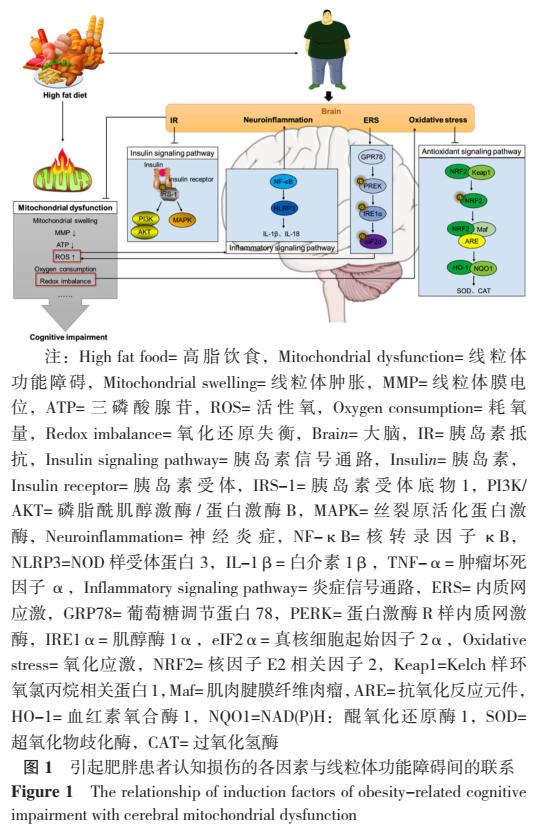
线粒体是调节细胞功能和生存的多种细胞内信号通路的重要参与者,笔者对椎间盘退变相关的线粒体分子机制进行综述,并讨论了将线粒体作为治疗椎间盘退变靶点的潜能。表明在椎间盘不利微环境下,椎间盘细胞发生线粒体功能障碍,功能障碍的线粒体通过调节氧化应激、细胞死亡、线粒体自噬、线粒体分裂/融合及其之间的相互串扰,引起椎间盘细胞死亡、基质稳态破坏和炎症反应,最终促进椎间盘退变的发生和进展。
本文作者
陈胜1,2 陈明珏2 林嗣雄2,3 曹惠玲2 邵增务1* 肖国芝2*
作者单位
1 华中科技大学同济医学院附属协和医院骨科,湖北 武汉,430022;2 南方科技大学医学院生物化学系,广东 深圳,518055;3 中山大学第一附属医院脊柱外科,广东 广州,510080\\
作者简介
[作者简介]陈胜(1993-)男,博士研究生。研究方向:椎间盘退变。
*[通信作者]邵增务(1962-)男,博士,教授。研究方向:骨与软组织肿瘤及椎间盘退变的临床和基础研究。
*[通信作者]肖国芝(1963-)男,博士,教授。研究方向:骨骼发育和骨骼疾病。
引用信息
陈胜, 陈明珏, 林嗣雄, 等. 线粒体功能障碍在椎间盘退变中的作用[J]. 生物骨科材料与临床研究, 2021, 18(3): 1-5.
[摘要]线粒体是调节细胞功能和生存的多种细胞内信号通路的重要参与者,在年龄相关的退行性疾病中扮演了至关重要的角色。椎间盘退变是一种年龄相关的退行性疾病,以细胞外基质降解加速、细胞丢失和炎症反应为特征。近年来,大量体内外研究报道称,线粒体功能障碍通过影响多种病理生理过程,包括氧化应激、炎症小体激活、线粒体自噬、细胞衰老、细胞死亡和线粒体分裂/融合,参与了椎间盘退变的进展。本文对椎间盘退变相关的线粒体分子机制进行综述,并讨论了将线粒体作为治疗椎间盘退变靶点的潜能。
[关键词]椎间盘;线粒体;氧化应激;凋亡;动力学
下腰痛(low back pain,LBP)一般是指位于下位肋骨边缘和臀部折痕之间的疼痛,严重影响了人们的生活质量,给社会经济造成了沉重的负担。据流行病学调查,LBP的平均总患病率为31.0%,终生患病率为38.9%[1],在伤残损失寿命年影响因素中排名第一[2]。对高收入国家疾病费用进行统计分析表明,治疗LBP的总花费与其他常见的花费巨大的疾病相当,如癌症、心血管疾病、自身免疫性疾病和精神疾病等[3]。椎间盘退变(intervertebral disc degeneration,IDD)与LBP的发生密切相关,被认为是导致LBP的主要影响因素之一[4]。因此,研究和阐明IDD的分子机制有助于LBP的治疗。在过去几十年里,研究者发现线粒体功能障碍可以导致细胞和器官功能失调,进而导致一系列人类疾病,揭示了线粒体在人类健康中的重要作用[5]。近年来,越来越多的研究发现,线粒体功能障碍参与了IDD的发生发展,线粒体在维持椎间盘稳态中具有重要作用[6-7]。因此,本文旨在结合相关的研究报道和最新的见解,对线粒体功能障碍相关的分子机制在IDD中的作用进行综述,并对将线粒体作为治疗椎间盘退变靶点进行了展望。
1 椎间盘的生物学特征
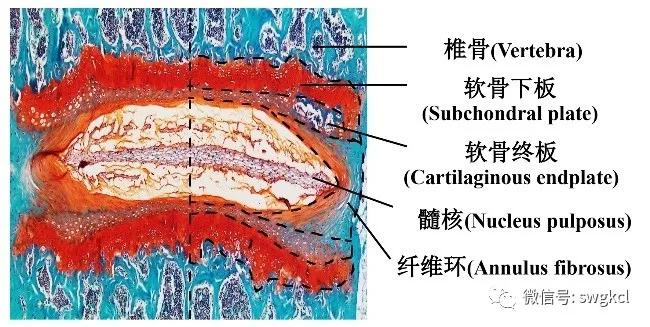
正常的椎间盘是一种纤维软骨样的结构,位于脊柱上下椎体之间,被肌肉和韧带包围[8]。在人体内,从C2-3到L5-S1共23块椎间盘,起着缓冲肌肉紧张和身体重量对脊柱施加压力的作用,为脊柱提供稳定性和多轴灵活性。椎间盘是一种乏血供的器官,主要由中央的髓核,外周的纤维环和上、下软骨终板(软骨终板与邻近的软骨下板在概念上容易混淆,见图1)三部分组成[9]。椎间盘的每一部分都有区域特异的原位椎间盘细胞(如髓核细胞、纤维环细胞等),这些细胞可以合成和分泌细胞外基质,参与椎间盘稳态的维持。由于椎间盘特殊的结构组成,椎间盘内呈现的是过度压力、高渗、低pH和营养缺乏的不利微环境。这些不利因素诱导椎间盘细胞衰老、促进细胞死亡、激活炎症反应、破坏细胞外基质分解/合成平衡,最终导致IDD。然而,其中的具体机制还没有完全阐明[10]。
2 线粒体的结构和功能
线粒体作为真核细胞的能量供应中心,在ATP和许多其他生物合成中间物的生产中起着关键作用,并参与构建了细胞应激反应中相互串扰的信号通路[5,11]。线粒体由组成和功能不同的线粒体外膜(mitochondrial outer membrane,MOM)和线粒体内膜(mitochondrial inner membrane,MIM)分为两个间隔,膜间隙(intermembrane space,IMS)和基质。MOM含有多种孔蛋白、膜转运蛋白和酶,这些成分为MOM内外的沟通奠定基础,也支持了其作为信号平台的诸多功能,如调节代谢、活性氧(reactive oxygen species,ROS)的产生和释放、Ca2+稳态、线粒体自噬和细胞死亡等[12]。MIM由于折叠多次形成了线粒体嵴结构,显著扩大了膜的面积。MIM主要含有调控氧化磷酸化和ATP合成的电子传递链(electron transport chain,ETC)蛋白复合物,以及转运蛋白和调节线粒体动力学的蛋白。IMS是位于MOM和MIM之间的间隙,参与了蛋白加工、蛋白降解以及MIM和细胞质之间代谢物的转运。线粒体基质也称为线粒体内腔,是MIM包绕形成的线粒体内部间隙。线粒体基质主要含有线粒体DNA、有机小分子、离子以及三羧酸循环和其他代谢通路的酶。
3 线粒体功能障碍与IDD
3.1 线粒体功能障碍导致的氧化应激在IDD中的作用
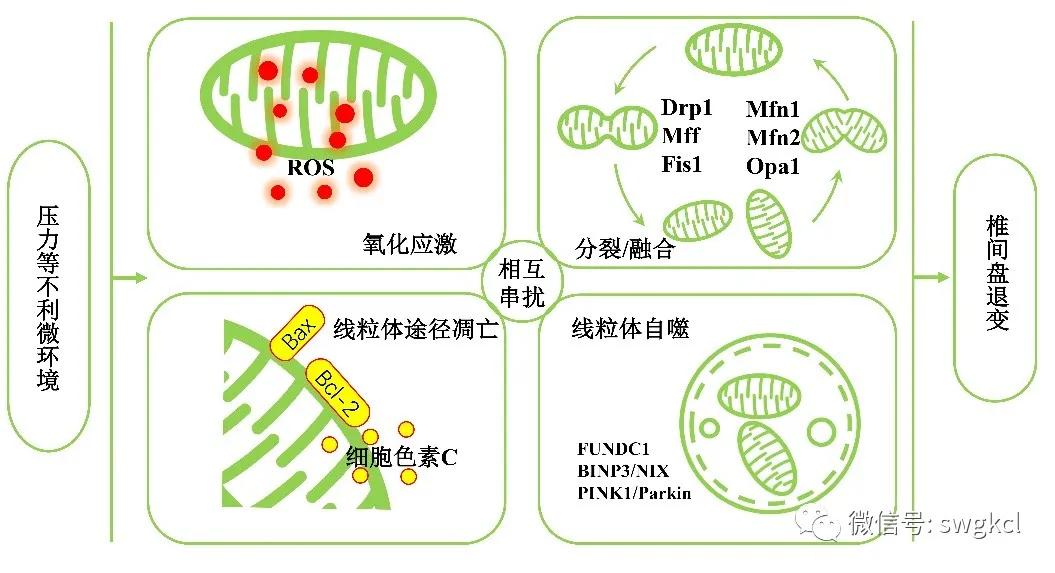
氧化应激是指细胞内ROS水平明显超过生理阈值的一种状态,在IDD的发生发展中起着重要作用[13]。ROS是一类含氧的高效化学反应性氧化剂,包括过氧化氢(H2O2)、超氧化物阴离子自由基(·O2-)、羟自由基(·OH)和单态氧(1O2)。在生理状态下,ROS的产生主要是线粒体由状态Ⅲ向状态Ⅳ转换中高氧的环境和高还原态的呼吸链使大量电子漏出并还原氧分子而形成的[14]。在椎间盘中,不利微环境可以导致椎间盘细胞内线粒体通透性转换孔(mitochondrial permeability transition pore,MPTP)开放、线粒体膜电位(mitochondrial membrane potential,MMP)降低,使得小分子物质大量进入线粒体内膜,引起线粒体肿胀、线粒体嵴消失、呼吸链受损,最终导致线粒体功能障碍。功能发生障碍的线粒体会漏出大量的电子,促使椎间盘细胞内ROS增加,过度增加的ROS反过来还会促进线粒体功能障碍,使ROS产生进一步增多,形成恶性循环[15]。目前已有报道,压力[16]、高糖[17]、高渗[18]和血清剥夺[19]等不利微环境都会导致椎间盘细胞内线粒体功能障碍,使椎间盘细胞内线粒体ROS过度增加。椎间盘内过度增加的ROS会对椎间盘细胞造成氧化应激损伤(见图2),可表现为:①细胞增殖减慢,细胞周期阻滞,衰老相关分泌表型(TNF-α、IL-1β、IL-6和IL-8等)表达升高,p53/p21和p16/Rb通路激活,细胞发生衰老[18];②NLRP3炎症小体激活,IL-1β释放增多,加剧炎症反应[20];③细胞发生线粒体动力学改变、死亡[21];④细胞外基质合成和分解平衡被破坏[15]。由此可见,在治疗IDD的过程中,我们可以通过改善线粒体功能,减少氧化应激损害。
3.2 线粒体途径的细胞凋亡在IDD中的作用
细胞凋亡是一种程序性细胞死亡方式,表现为细胞皱缩、核固缩、DNA断裂和凋亡小体形成。细胞凋亡主要分为线粒体途径的细胞凋亡(也称为内源性途径的细胞凋亡)和外源性途径的细胞凋亡[22]。线粒体途径的细胞凋亡是不利微环境导致椎间盘细胞死亡的一种主要死亡方式[16]。在各种刺激作用下,线粒体上的抗凋亡Bcl-2蛋白表达下降,促凋亡唯BH3域蛋白活化,促凋亡Bax和Bak蛋白寡聚化,线粒体通透性转换孔开放,细胞色素C释放。从线粒体释放出的细胞色素C与凋亡蛋白酶激活因子1结合并诱导凋亡复合体形成,随后caspase-9被招募激活并进一步激活caspase-3/7,最终导致细胞凋亡。文献报道,椎间盘内异常的应力[23]、糖基化终末产物[24]、高糖[25]和炎症[26]等都会造成线粒体途径的细胞凋亡(见图2)。笔者前期的实验也证明了线粒体途径的细胞凋亡在椎间盘退变中起着重要作用,但是同时还发现抑制凋亡并不能很好地抑制不利刺激因素造成的椎间盘细胞死亡[27]。近期的一些实验证据表明,椎间盘退变过程中还存在其他细胞死亡方式,包括坏死性凋亡[28]、焦亡[29]和铁死亡[30]。因此,找到能联合调控多种细胞死亡的靶点可能是治疗IDD的一种有效方法。
3.3 线粒体自噬在IDD中的作用
线粒体自噬指在不利因素刺激下,线粒体受到损伤、功能发生障碍,细胞中的自噬小体特异性地识别和包被受损的线粒体,并与溶酶体融合形成自噬溶酶体,最终被降解的过程(见图2)。线粒体自噬根据自噬体识别途径的不同可分为:①由泛素介导的自噬,主要由PINK1/Parkin通路参与;②由线粒体自噬受体蛋白介导的自噬,主要由BINP3/NIX、FUNDC1蛋白参与[31]。线粒体自噬在IDD的发生发展中起到双刃剑的作用:一方面,适当的线粒体自噬激活有利于椎间盘细胞稳态的维持。研究发现,血红素氧合酶-1可以通过激活线粒体自噬抑制人髓核细胞发生的复制性衰老[32],尿石素A可以通过激活线粒体自噬抑制叔丁基过氧化氢诱导的髓核细胞凋亡[33]。另一方面,线粒体自噬的过度激活可促进IDD的进展。笔者的研究发现,过度的压力可以通过激活线粒体自噬导致髓核细胞发生衰老[34]。Xu等[35]的实验结果表明,氧化应激可以通过提高线粒体自噬水平诱导人髓核细胞发生凋亡。有研究证实,氧化应激中大量的ROS可以通过PI3K-AKT-mTOR和MAPK通路过度激活自噬[36]。而在过度的自噬中,caspase-8激活上调以及凋亡蛋白抑制因子降解增多可诱导细胞凋亡的产生[37]。因此,在未来的实验中,笔者需要深入探讨线粒体自噬相关的分子机制,并对其做到精准调控,这将有助于加强对IDD病因的理解和相关治疗靶点的开发。
3.4 线粒体分裂/融合失衡在IDD中的作用
线粒体是一个高度活跃的细胞器,在环境刺激下会发生分裂、融合和自噬等变化,这些过程统称为线粒体动力学[38]。其中,线粒体分裂和融合失衡在心血管疾病、肿瘤等多种疾病的发生发展中起着重要作用[39]。研究已证实有许多蛋白介导线粒体分裂和融合的过程,Drp1、Mff和Fis1蛋白主要参与线粒体分裂,Mfn1、Mfn2和Opa1蛋白主要参与线粒体融合[40]。有研究报道,过度的压力可导致髓核细胞线粒体分裂/融合失衡,表现为线粒体融合相关蛋白Mfn1、Mfn2和Opa1表达降低,线粒体分裂相关蛋白Drp1、Mff和Fis1表达升高,最终引起线粒体功能障碍[41]。Madhu等[7]发现在低氧条件下,髓核细胞Drp1蛋白表达升高,线粒体分裂增加。笔者最近的研究也发现Drp1介导的线粒体分裂参与了氧化低密度脂蛋白导致的纤维环细胞凋亡[42]。Chen等[43]报道Mfn2在人退变的椎间盘组织中表达降低,过表达Mfn2可以通过调节自噬缓解氧化应激对髓核细胞造成的损伤、延缓IDD。这些证据表明,压力等导致的椎间盘细胞线粒体分裂/融合失衡,可通过调节氧化应激、凋亡和自噬,促进IDD的进展(见图2)。椎间盘内其他不利微环境是否也可以通过影响线粒体分裂/融合平衡进而引起IDD目前还不清楚,需要深入研究。
4 以线粒体为靶点治疗IDD的展望
目前,IDD的治疗主要包括卧床休息、物理治疗、药物对症治疗和手术治疗,这些治疗手段都不能从根本上逆转退变的椎间盘组织。根据线粒体功能障碍促进IDD进展的相关分子机制,可以考虑从以下几个方面进行治疗干预:①抑制过多的线粒体ROS。有研究报道TIGAR分子可以通过提高NADPH/NADP+和还原型/氧化型谷胱甘肽的比例抑制髓核细胞线粒体ROS产生,减轻氧化应激对髓核细胞的损伤[44]。除了相关药物开发,还可以考虑将线粒体ROS清除剂和生物材料进行结合,实现精准、高效和长期的椎间盘内ROS清除效果。②抑制线粒体途径的凋亡。例如,淫羊藿苷可以通过升高Bcl-2表达、降低Bax表达抑制氧化应激诱导的髓核细胞凋亡,缓解IDD进展[45]。③调节线粒体自噬。有研究发现褪黑素可以通过激活线粒体自噬抑制髓核细胞凋亡,抑制细胞外基质分解[46]。此外,可以针对凋亡和自噬之间串扰的相关靶点开发药物,实现联合调控,这可能是修复IDD的一个良好策略。④调节线粒体分裂/融合。例如,米托蒽醌可以改善过度压力造成的线粒体分裂/融合失衡,进而治疗IDD[41]。⑤其他如干细胞治疗,通过细胞间线粒体转移提供正常的线粒体或干细胞分泌外泌体、细胞因子等改善线粒体功能障碍[47-48]。总之,基于线粒体功能在椎间盘稳态中的重要作用,将线粒体作为靶点治疗IDD可能会成为一个非常具有前景的研究方向。
5 总结
在椎间盘不利微环境下,椎间盘细胞发生线粒体功能障碍,功能障碍的线粒体通过调节氧化应激、细胞死亡、线粒体自噬、线粒体分裂/融合及其之间的相互串扰,引起椎间盘细胞死亡、基质稳态破坏和炎症反应,最终促进IDD的发生和进展。在未来的研究中,对IDD中相关线粒体分子机制的深入探讨及针对靶点进行治疗干预,将会使我们对IDD疾病的认识进入一个新阶段。
参考文献
[1]Hoy D, Bain C, Williams G, et al. A systematic review of the global prevalence of low back pain[J]. Arthritis Rheum, 2012, 64(6): 2028-2037.
[2]Collaborators GD. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017[J]. Lancet, 2018, 392(10159): 1789-1858.
[3]Maher C, Underwood M, Buchbinder R. Non-specific low back pain[J]. Lancet, 2017, 389(10070): 736-747.
[4]Yang S, Zhang F, Ma J, et al. Intervertebral disc ageing and degeneration: The antiapoptotic effect of oestrogen[J]. Ageing Res Rev, 2020, 57: 100978.
[5]Bornstein R, Gonzalez B, Johnson SC. Mitochondrial pathways in human health and aging[J]. Mitochondrion, 2020, 54: 72-84.
[6]Kang L, Xiang Q, Zhan S, et al. Restoration of autophagic flux rescues oxidative damage and mitochondrial dysfunction to protect against intervertebral disc degeneration[J]. Oxid Med Cell Longev, 2019, 2019: 7810320.
[7]Madhu V, Boneski PK, Silagi E, et al. Hypoxic regulation of mitochondrial metabolism and mitophagy in Nucleus pulposus cells is dependent on HIF-1alpha-BNIP3 axis[J]. J Bone Miner Res, 2020, 35(8): 1504-1524.
[8]Lyu FJ, Cheung KM, Zheng Z, et al. IVD progenitor cells: A new horizon for understanding disc homeostasis and repair[J]. Nat Rev Rheumatol, 2019, 15(2): 102-112.
[9]Huang YC, Urban JP, Luk KD. Intervertebral disc regeneration: Do nutrients lead the way?[J]. Nat Rev Rheumatol, 2014, 10(9): 561-566.
[10]Chen S, Liu S, Zhao L, et al. Heme oxygenase-1-mediated autophagy protects against oxidative damage in rat nucleus pulposus-derived mesenchymal stem cells[J]. Oxid Med Cell Longev, 2020, 2020: 9349762.
[11]Shanmughapriya S, Langford D, Natarajaseenivasan K. Inter and intracellular mitochondrial trafficking in health and disease[J]. Ageing Res Rev, 2020, 62: 101128.
[12]Deus CM, Yambire KF, Oliveira PJ, et al. Mitochondria-lysosome crosstalk: From physiology to neurodegeneration[J]. Trends Mol Med, 2020, 26(1): 71-88.
[13]Nasto LA, Robinson AR, Ngo K, et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration[J]. J Orthop Res, 2013, 31(7): 1150-1157.
[14]Grasso D, Zampieri L X, Capeloa T, et al. Mitochondria in cancer[J]. Cell Stress, 2020, 4(6): 114-146.
[15]Feng C, Yang M, Lan M, et al. ROS: Crucial intermediators in the pathogenesis of intervertebral disc degeneration[J]. Oxid Med Cell Longev, 2017, 2017: 5601593.
[16]Chen S, Zhao L, Deng X, et al. Mesenchymal stem cells protect nuc-leus pulposus cells from compression-induced apoptosis by inhibiting the mitochondrial pathway[J]. Stem Cells Int, 2017, 2017: 9843120.
[17]Jiang Z, Lu W, Zeng Q, et al. High glucose-induced excessive reactive oxygen species promote apoptosis through mitochondrial damage in rat cartilage endplate cells[J]. J Orthop Res, 2018, 36(9): 2476-2483.
[18]Xu J, Li H, Yang K, et al. Hyper-osmolarity environment-induced oxidative stress injury promotes nucleus pulposus cell senescence in vitro[J]. Biosci Rep. 2019, 39(9): BSR20191711.
[19]Chen JW, Ni BB, Zheng XF, et al. Hypoxia facilitates the survival of nucleus pulposus cells in serum deprivation by down-regulating excessive autophagy through restricting ROS generation[J]. Int J Biochem Cell Biol, 2015, 59: 1-10.
[20]Zhao Y, Qiu C, Wang W, et al. Cortistatin protects against intervertebral disc degeneration through targeting mitochondrial ROS-dependent NLRP3 inflammasome activation[J]. Theranostics, 2020, 10(15): 7015-7033.
[21]Ma K, Chen S, Li Z, et al. Mechanisms of endogenous repair failure during intervertebral disc degeneration[J]. Osteoarthritis Cartilage, 2019, 27(1): 41-48.
[22]Kist M, Vucic D. Cell death pathways: Intricate connections and disease implications[J]. EMBO J, 2021: e106700.
[23]Fu F, Bao R, Yao S, et al. Aberrant spinal mechanical loading stress triggers intervertebral disc degeneration by inducing pyroptosis and nerve ingrowth[J]. Sci Rep, 2021, 11(1): 772.
[24]Hu Y, Shao Z, Cai X, et al. Mitochondrial pathway is involved in advanced glycation end products-induced apoptosis of rabbit annulus fibrosus cells[J]. Spine (Phila Pa 1976), 2019, 44(10): E585-E595.
[25]Guo MB, Wang DC, Liu HF, et al. Lupeol against high-glucose- induced apoptosis via enhancing the anti-oxidative stress in rabbit nucleus pulposus cells[J]. Eur Spine J, 2018, 27(10): 2609-2620.
[26]Shen J, Xu S, Zhou H, et al. IL-1beta induces apoptosis and autophagy via mitochondria pathway in human degenerative nucleus pulposus cells[J]. Sci Rep, 2017, 7: 41067.
[27]Chen S, Lv X, Hu B, et al. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death[J]. Apoptosis, 2017, 22(5): 626-638.
[28]Zhang QX, Guo D, Wang FC, et al. Necrosulfonamide (NSA) protects intervertebral disc degeneration via necroptosis and apoptosis inhibition[J]. Eur Rev Med Pharmacol Sci, 2020, 24(5): 2683-2691.
[29]Zhao K, An R, Xiang Q, et al. Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration[J]. Cell Prolif, 2021, 54(1): e12941.
[30]Zhang X, Huang Z, Xie Z, et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4[J]. Free Radic Biol Med, 2020, 160: 552-565.
[31]Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy[J]. EMBO J, 2021: e104705.
[32]Yi W, Lan H, Wen Y, et al. HO-1 overexpression alleviates senescence by inducing autophagy via the mitochondrial route in human nucleus pulposus cells[J]. J Cell Physiol, 2020, 235(11): 8402-8415.
[33]Lin J, Zhuge J, Zheng X, et al. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway[J]. Free Radic Biol Med, 2020, 150: 109-119.
[34]Huang D, Peng Y, Li Z, et al. Compression-induced senescence of nucleus pulposus cells by promoting mitophagy activation via the PINK1/PARKIN pathway[J]. J Cell Mol Med, 2020, 24(10): 5850-5864.
[35]Xu WN, Zheng HL, Yang RZ, et al. Mitochondrial NDUFA4L2 attenuates the apoptosis of nucleus pulposus cells induced by oxidative stress via the inhibition of mitophagy[J]. Exp Mol Med, 2019, 51(11): 1-16.
[36]Gao Q. Oxidative stress and autophagy[J]. Adv Exp Med Biol, 2019, 1206: 179-198.
[37]Yan X, Zhou R, Ma Z. Autophagy-cell survival and death[J]. Adv Exp Med Biol, 2019, 1206: 667-696.
[38]Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury[J]. Angiogenesis, 2020, 23(3): 299-314.
[39]Chan DC. Mitochondrial dynamics and its involvement in disease[J]. Annu Rev Pathol, 2020, 15: 235-259.
[40]Forte M, Schirone L, Ameri P, et al. The role of mitochondrial dynamics in cardiovascular diseases[J]. Br J Pharmacol, 2020. [Epub ahead of print].
[41]Kang L, Liu S, Li J, et al. The mitochondria-targeted anti-oxidant MitoQ protects against intervertebral disc degeneration by ameliorating mitochondrial dysfunction and redox imbalance[J]. Cell Prolif, 2020, 53(3): e12779.
[42]Wu W, Jing D, Huang X, et al. Drp1-mediated mitochondrial fission is involved in oxidized low-density lipoprotein-induced AF cell apoptosis[J]. J Orthop Res. 2020. Epub ahead of print.
[43]Chen Y, Lin J, Chen J, et al. Mfn2 is involved in intervertebral disc degeneration through autophagy modulation[J]. Osteoarthritis Cartilage, 2020, 28(3): 363-374.
[44]Jiang LB, Cao L, Ma YQ, et al. TIGAR mediates the inhibitory role of hypoxia on ROS production and apoptosis in rat nucleus pulposus cells[J]. Osteoarthritis Cartilage, 2018, 26(1): 138-148.
[45]Hua W, Li S, Luo R, et al. Icariin protects human nucleus pulposus cells from hydrogen peroxide-induced mitochondria-mediated apoptosis by activating nuclear factor erythroid 2-related factor 2[J]. Biochim Biophys Acta Mol Basis Dis, 2020, 1866(1): 165575.
[46]Chen Y, Wu Y, Shi H, et al. Melatonin ameliorates intervertebral disc degeneration via the potential mechanisms of mitophagy induction and apoptosis inhibition[J]. J Cell Mol Med, 2019, 23(3): 2136-2148.
[47]Mohammadalipour A, Dumbali SP, Wenzel PL. Mitochondrial transfer and regulators of mesenchymal stromal cell function and therapeutic efficacy[J]. Front Cell Dev Biol, 2020, 8: 603292.
[48]Xia C, Zeng Z, Fang B, et al. Mesenchymal stem cell-derived exosomes ameliorate intervertebral disc degeneration via anti-oxidant and anti-inflammatory effects[J]. Free Radic Biol Med, 2019, 143: 1-15.
来源:生物骨科材料与临床研究
声明:此文内容及图片由供稿单位提供,仅供学习交流,不代表骨科在线观点。
版权声明:CosMeDna所有作品(图文、音视频)均由用户自行上传分享,仅供网友学习交流。若您的权利被侵害,请联系删除!
本文链接://www.cosmedna.com/article/589573554.html